 |
Case Report
Eosinophilic granulomatosis with polyangiitis: Not all infiltrates are pneumonia
1 OMS-4, Burrell College of Osteopathic Medicine, Las Cruces, NM, USA
Address correspondence to:
Tyler Everett Wilson
OMS-4, Burrell College of Osteopathic Medicine, Las Cruces, NM,
USA
Message to Corresponding Author
Article ID: 100055Z09TW2020
Access full text article on other devices

Access PDF of article on other devices


How to cite this article
Wilson TE, Tersakyan S. Eosinophilic granulomatosis with polyangiitis: Not all infiltrates are pneumonia. J Case Rep Images Med 2020;6:100055Z09TW2020.ABSTRACT
Introduction: In the presence of pulmonary infiltrates, eosinophilic granulomatosis with polyangiitis may be hidden behind more obvious diagnoses. The disease can take up to several decades to progress to its end stages. However, if caught early, recovery and remission are possible.
Case Report: We present the case of a 55-year-old Caucasian female with a 20-year history of asthma along with multiple episodes of pneumonia. Lab results at the time of presentation showed marked eosinophilia in her blood, which prompted further investigation and the possible differential diagnoses. Additionally, we discuss the various diagnostic tests and management strategies that were employed by her medical team.
Conclusion: With the initial stages of eosinophilic granulomatosis with polyangiitis frequently mimicking more common pulmonary diseases, the diagnosis is often overlooked and delayed. As such, eosinophilic granulomatosis with polyangiitis should be considered as a possible differential diagnosis, despite seemingly clear signs of a more common disease.
Keywords: Asthma, Eosinophilia, Eosinophilic granulomatosis with polyangiitis, Pneumonia
Introduction
Eosinophilic granulomatosis with polyangiitis (EGPA) is a multisystem vasculitis affecting small and medium sized arteries. Although multiple organ systems may be involved, EGPA is characteristically associated with pulmonary involvement, which manifests as asthma with peripheral eosinophilia [1]. The disorder classically presents in three distinct, successive phases: prodromal, eosinophilic, and vasculitic phases [2]. The prodromal phase typically occurs first and presents with atopy and/or asthma. An eosinophilic phase subsequently develops and is characterized by peripheral eosinophilia with infiltration of organs, such as the lungs [2]. Finally, a vasculitic phase occurs with systemic vasculitis of small and medium vessels. This phase is associated with vascular and extravascular granulomas and typically presents with constitutional symptoms, such as fever, weight loss, and fatigue [2],[3]. Here, we present a patient with an episode of shortness of breath that ultimately leads to the diagnosis of EGPA.
Case Report
A 55-year-old female presented to the emergency room for worsening dyspnea with associated intractable nonproductive cough. She generally felt poor which had accelerated over the past week. She noted a fever recorded at 38.7 °C with one episode of nausea and vomiting. The patient denied having chest pain, abdominal pain, headache, dizziness, or light-headedness.
The patient had a tonsillectomy at age three and had her wisdom teeth removed as a child. In high school, she fractured her finger in multiple sites while playing basketball. She had a lymph node biopsy ten years ago with no significant findings. She also had a colonoscopy with polypectomy four years ago. She suffers from allergic rhinitis, asthma, recurrent bronchitis, migraines, and hyperlipidemia. She also experienced an episode of drug-induced idiopathic thrombocytopenic purpura (ITP) after being treated with levofloxacin.
The patient lived with her husband and two children, and she worked at an energy conservation laboratory. She exercised for 40 minutes six times per week and reported minimal stress in her life. She admitted to drinking two glasses of wine per week and denied any history of smoking. She did not admit to any recent travel or pet or insect exposures. Her mother has a history of osteoporosis, hyperlipidemia, dementia, chronic obstructive pulmonary disease (COPD), and pulmonary fibrosis. The patient’s father has a history of acute lymphoblastic leukemia and hyperlipidemia. Her sister has obesity, migraines, and melanoma.
Her current medications included an albuterol 90 mcg actuation inhaler to be used as needed, benzonatate 100 mg capsule three times per day as needed, ergocalciferol 50,000 unit capsule one time per week, fluticasone propionate/formoterol fumarate 230-21 mcg actuation inhaler, fluticasone propionate 50 mcg actuation nasal spray two sprays into each nostril one time per day, and montelukast 10 mg tablet one time per day per mouth. Her medication allergies included budesonide-formoterol and quinolones.
On examination, the temperature was 37.5 °C, the heart rate 108 beats per minute, the blood pressure 153/92 mmHg, and the oxygen saturation 94% on ambient air. She did have an intractable cough. There was no respiratory distress and no wheezes or rales were heard. There were no other significant findings on exam.
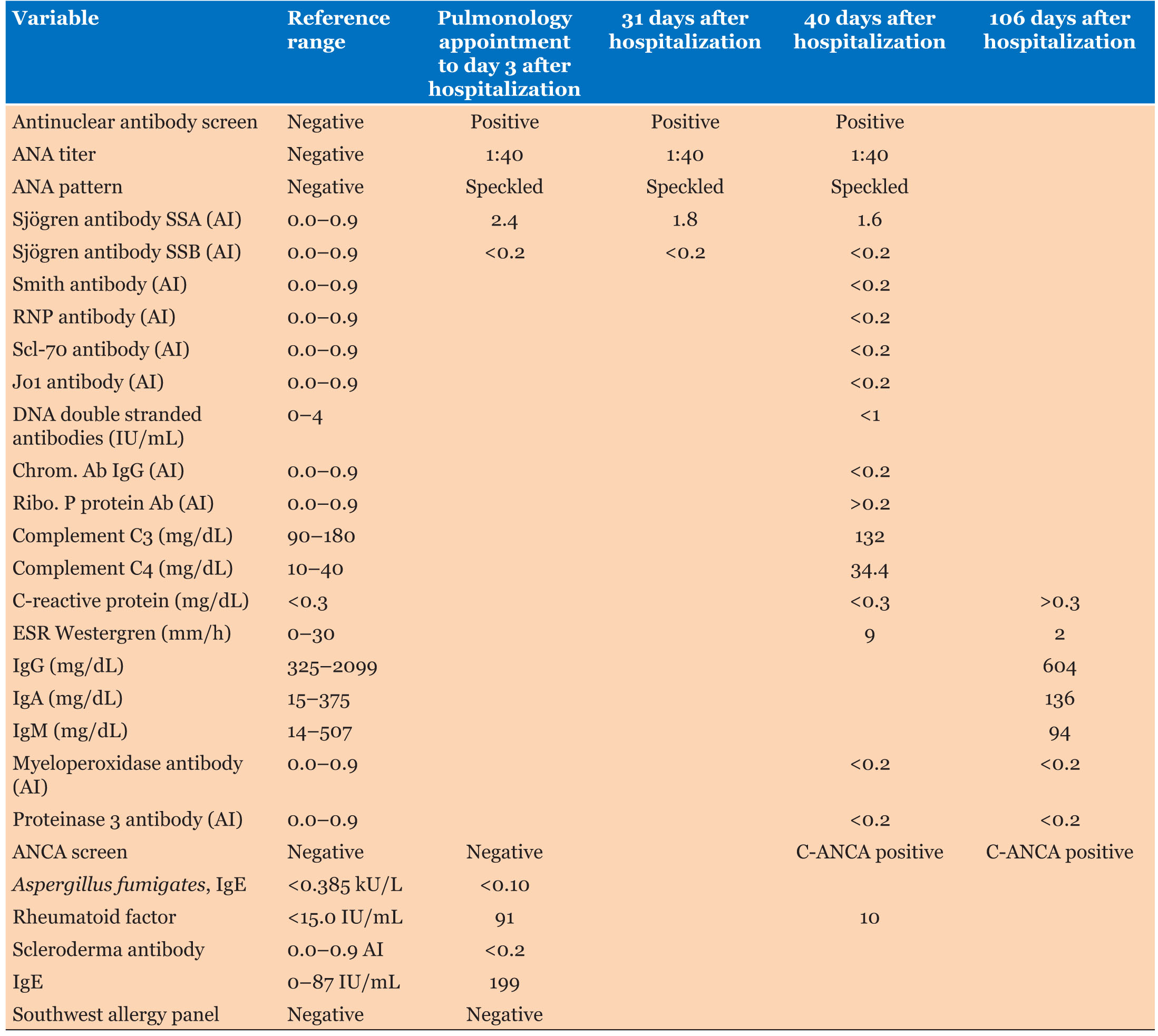
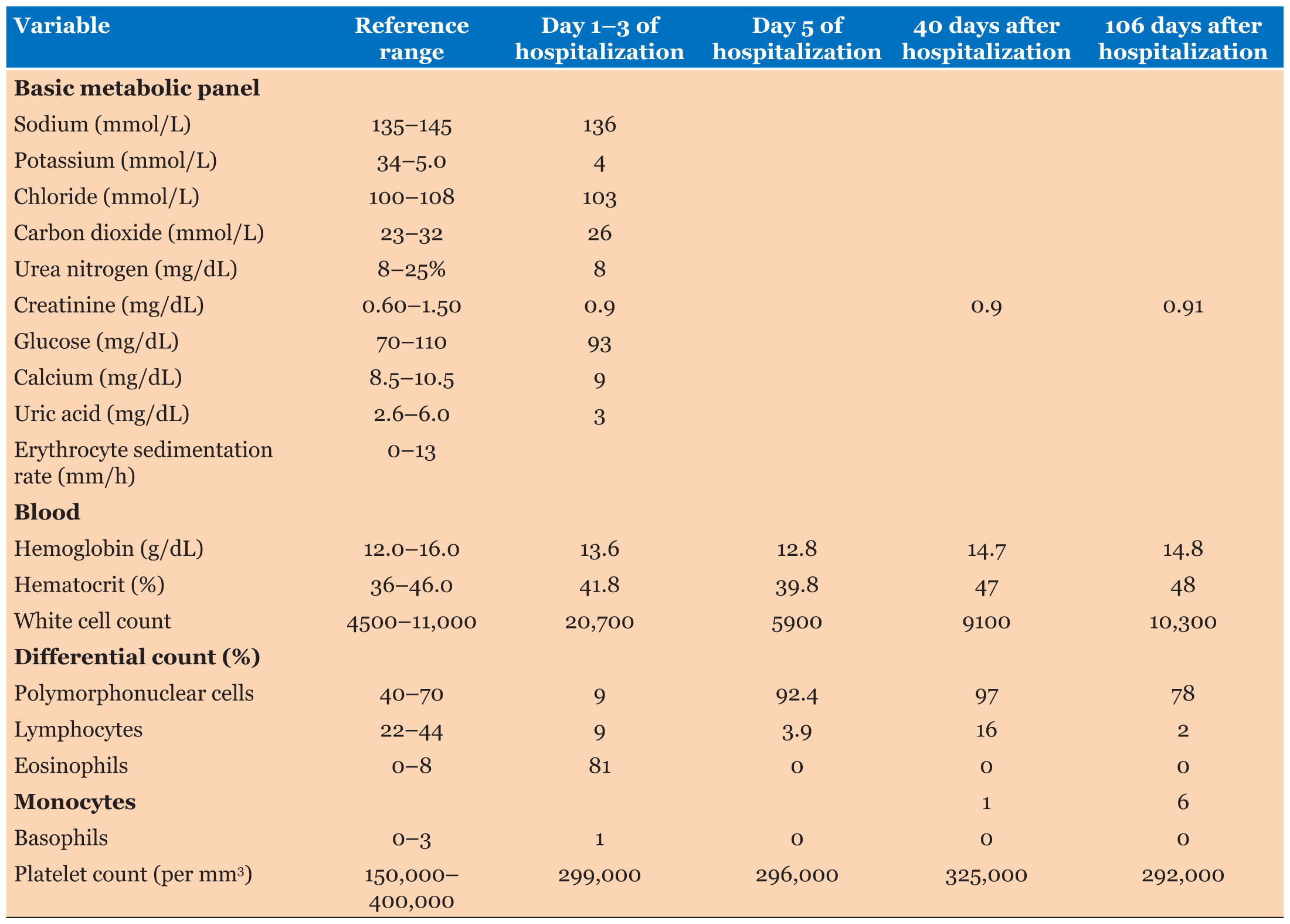
Labs showed significant increase in her white blood cell count at 20.7 with 81% eosinophils along with normal chemistries (Table 1 and Table 2).
Radiologist: Chest X-ray showed diffuse irregular opacities throughout both lungs, left lung worse than right, that were grossly unchanged from one day prior.
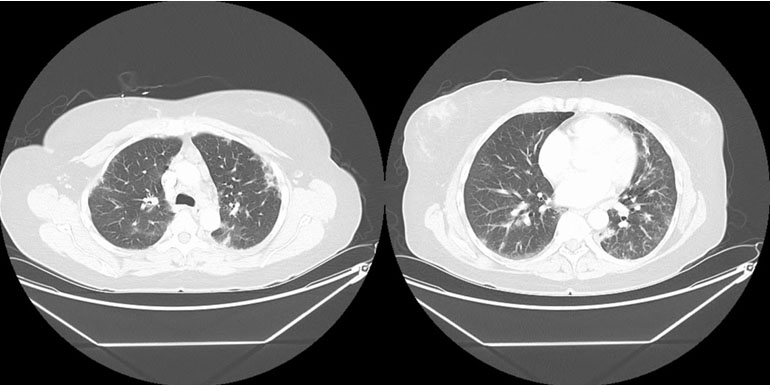
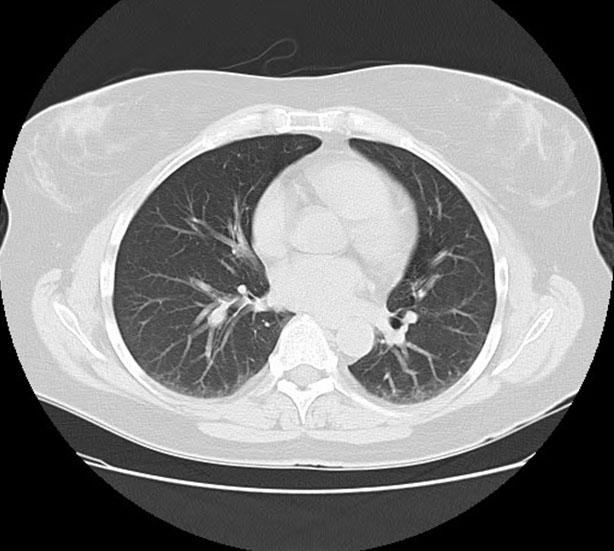
Radiologist: Computed tomography (CT) of the chest with contrast (Figure 1) showed multifocal groundglass opacities that were re-demonstrated in both lungs. These were weighted in the lower lobes, left worse than right. Additional lymphadenopathy included perivascular lymph nodes with one selectively measuring 0.6 cm short axis. Multiple lymph nodes demonstrated abnormal-rounded morphology. Bronchoscopy data might be informative.
The patient was then given a hydrocodone 5-325 mg tablet to control her cough. She was also placed on oxygen which seemed to help, but she had not been hypoxic. She was seen by pulmonary medicine, which ordered prednisone 70 mg tablet. The patient was then admitted to the medical floor for intravenous (IV) steroids and endobronchial ultrasound (EBUS) bronchoscopy.
Pathologist: EBUS bronchoscopy biopsy was negative for granulomas. Biopsy at lymph node 4R did show some eosinophils.
After being discharged from the hospital, the patient was referred to rheumatology. The rheumatologist ordered an anti-neutrophil cytoplasmic antibody (ANCA) test, and the test was positive. The patient was then started on azathioprine 100 mg tablet daily and told to continue prednisone 10 mg daily.
One day before the presentation, pulmonology workup done in the outside hospital did show positive rheumatoid factor (RF) and an immunoglobulin E (IgE) level of 199 IU/mL (reference range, 0–87) but negative antineutrophil cytoplasmic antibodies (ANCA) (Table 1).
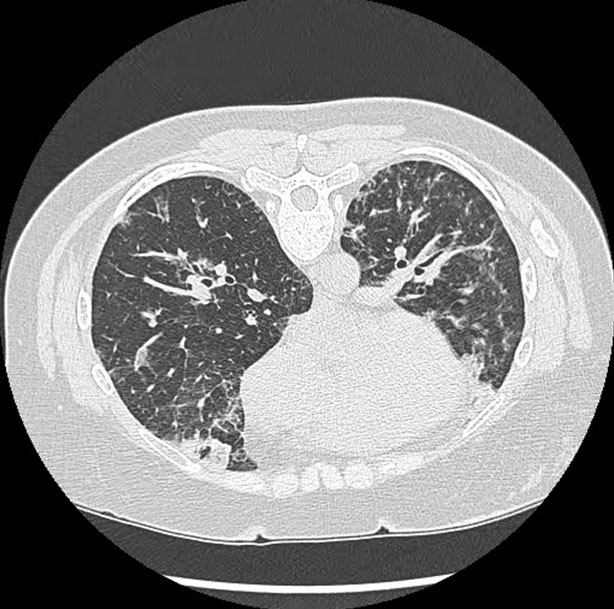
Radiologist: the chest CT without intravenous contrast (Figure 2) showed bilateral peripheral areas of consolidation and ground-glass opacities which persisted on the prone images. Septal thickening was present, primarily in the lung bases. Diffuse centrilobular nodules were noted as well. There was also apparent mediastinal adenopathy.
Two days before the presentation, the patient was seen by pulmonology. She reported feeling chills, myalgias, and a fever. She also noted cough, shortness of breath, and wheezing. On examination, the temperature was 37.3 °C, the heart rate 104 beats per minute, the blood pressure 134/80 mmHg, and the oxygen saturation 90% while the patient was breathing ambient air. Her respiratory effort and breath sounds were normal.
Pulmonologist: A chest X-ray from one week prior showed heart size and pulmonary vascularity within normal limits. There were multifocal ill-defined infiltrates seen in the right upper lobe, left upper lobe, and left lower lobe without definite peribronchial interstitial thickening to suggest reactive airways disease. No associated pleural effusions were identified. There may had been some bronchiectatic changes in the lingular region. Pulmonary function tests (PFTs) and chest CT were clinically warranted.
Pulmonology continued empiric therapy with albuterol for concerns of wheezing and associated cough. The patient was placed on antitussive medications for symptom relief with benzonatate. A workup that included tests for allergic bronchopulmonary aspergillosis (ABPA) IgE and a Southwest Allergy Panel was ordered. Additional blood work was ordered that included allergen Aspergillus fumigatus IgE, allergen Southwest Panel, IgE, rheumatoid factor, SCL-70, Sjögren’s antibodies (SSA, SSB), and antinuclear antibodies (Table 1). Additionally, her PFTs did not show any signs of asthma. Reversible pulmonary causes with concerns of bronchiectasis were to be evaluated with a chest CT without intravenous contrast. She was not started on any antibiotics due to poor previous responses. The patient was then advised to go to the emergency room if she experienced any worsening symptoms. A follow-up visit with pulmonology was scheduled for one week to decide if further workup or treatment was necessary.
Approximately three weeks before the presentation, the patient established with a new primary care physician to address continued concerns about her asthma and allergies. The patient did have a low-grade fever and was found to have eosinophilia and infiltrates when evaluated in the urgent care. The patient was treated with doxycycline, but her symptoms did not improve. She was also referred to pulmonology for further evaluation of her asthma and chronic cough.
Approximately 15 months before the presentation, the patient experienced worsening of her asthma symptoms. She had multiple evaluations in the urgent care and emergency room for concerns of acute exacerbations and pneumonia. The patient also had multiple labs done, which showed persistent eosinophilia.
Approximately 10 and 12 years before the presentation, she was evaluated for nonspecific lymphadenopathy, which was inconclusive. She was also considered and worked up for thrombocytopenia through her oncologist.
Approximately 20 years before the presentation, the patient began to be treated by her primary care provider for asthma related symptoms.
The patient’s differential included eosinophilic granulomatosis with polyangiitis, bronchopulmonary aspergillosis, chronic eosinophilic pneumonia, and Sjögren syndrome (SS). The patient’s history of asthma, complete blood count (CBC) with a differential of >10% eosinophilia, recurrent non-fixed pulmonary infiltrates, and blood vessel biopsy demonstrating extravascular eosinophils confirmed the diagnosis of eosinophilic granulomatosis with polyangiitis.
Diagnosis: Eosinophilic granulomatosis with polyangiitis.
Discussion
Differential diagnosis
Evaluating a patient with an extensive history of chronic cough and negative workup can be difficult. Although the patient carried the diagnosis of asthma for several years, the progressive nature of her disease, along with the subsequent finding of eosinophilia, narrowed the differential diagnosis. When considering the patient’s marked 81% eosinophilia (Table 2), several categories of differential diagnoses must be considered: infectious diseases, medications, hematologic or oncologic disorders, immune dysregulation, and allergic disorders [4],[5]. After review of the patient’s presentation, and with a focus on respiratory involvement without improvement on antibiotics, the most likely differential diagnoses can be simplified to pulmonary eosinophilic disorders. With overlapping features in presentation, imaging, and lab values, the most likely diagnoses that were considered include eosinophilic granulomatosis with polyangiitis, bronchopulmonary aspergillosis, chronic eosinophilic pneumonia, and SS. Further workup and consideration of the patient’s constitutional symptoms raised most suspicion for ANCA-associated vasculitis including eosinophilic granulomatosis with polyangiitis [6].
Allergic bronchopulmonary aspergillosis
The International Society for Human and Animal Mycology (ISHAM) has outlined guidelines for the diagnosis of ABPA. Such diagnostic criteria include presentation with one of the following predisposing conditions: asthma or cystic fibrosis [7]. Although cases of ABPA without these predisposing conditions have been reported, studies show that this is an infrequent occurrence [8],[9]. Additional mandatory diagnostic criteria for ABPA include a positive aspergillus skin test (or detectable IgE levels against A. fumigatus), and elevated total serum IgE > 1000 IU/mL. It is important to note that the diagnosis may still be made with an IgE < 1000 IU/mL if all other criteria are met. Further, at least two of the following minor criteria must be present for diagnosis: precipitating serum antibodies to A. fumigatus, radiographic pulmonary opacities consistent with ABPA, and a current or previous history of an eosinophil count greater than 500 cells/mL in glucocorticoid naïve patients [7].
The diagnosis of ABPA was entertained due to the patient’s recurrent asthma exacerbations, eosinophilia, pulmonary consolidation with ground glass opacities, and bronchiectatic changes. Allergic bronchopulmonary aspergillosis was further supported after the patient completed a course of antibiotics without significant improvement, along with the lack of improvement in eosinophilia with prednisone. On further workup, however, several key factors helped rule this out as a possible diagnosis. These factors include, but are not limited to, negative Aspergillus antibodies, IgE < 1000 without meeting all other diagnostic criteria (Table 1), and lack of bronchial wall thickening on imaging (Figure 1). This diagnosis was effectively ruled out at the time of these findings.
Chronic eosinophilic pneumonia
Coming to the diagnosis of chronic eosinophilic pneumonia (CEP) requires combining clinical presentation with imaging or bronchoalveolar lavage. Laboratory values and lung biopsy also aid in making the diagnosis but are often not required [10]. Chronic eosinophilic pneumonia typically presents in women nonsmokers with subacute onset of cough, fever, progressive dyspnea, wheezing, weight loss, and night sweats [10],[11]. Workup reveals key features including characteristic peripheral opacities that are localized to the mid or upper lung zones without pleural effusion [11],[12]. Although less common, ground glass opacities are occasionally found in CEP as well [11]. Additionally, bronchoalveolar lavage typically demonstrates >25% eosinophilia and labs reveal a peripheral eosinophilia with a mean differential of 32.3% [11],[13],[14]. Other labs may reveal elevated IgE with a mean of 506 IU/mL, elevated C-reactive protein, and high sedimentation rate [15].
This differential was first considered by pulmonology when the patient was evaluated for chronic cough with CEP-like presentation: dyspnea, chills, fever, wheezing, and eosinophilia. Additionally, the patient’s eosinophilia initially did not improve with prednisone and her infiltrates did not improve with antibiotics. The patient’s chest X-ray also showed characteristic findings, which included multifocal, predominantly upper peripheral ill-defined infiltrates without definite peribronchial interstitial thickening. This differential was also suggested by radiology when a CT without contrast revealed findings that are highly suggestive of CEP: bilateral peripheral areas of consolidation and ground-glass opacities (Figure 2). On further workup, bronchoscopy revealed eosinophilia with an absence of infection, further supporting CEP as a possible diagnosis. With many overlapping features, the patient was referred to rheumatology, at which time ACNA was re-ordered. Although negative at admission, the patient’s c-ANCA came back positive 40 days after her hospitalization (Table 1). This pattern of late ANCA positivity made EGPA more likely, since ANCA is typically only positive in 40–60% of EGPA patients [16].
Sjögren’s syndrome
Sjögren’s syndrome is a chronic autoimmune disorder that typically presents with decreased lacrimal and salivary gland function. With no single diagnostic test, SS is a clinical diagnosis of exclusion that is supported by the presence of characteristic clinical and laboratory features. After ruling out other possible causes for presentation, several criteria can be used to suggest a diagnosis of SS. Such criteria include objective findings of ocularoral dryness, glandular parenchymal abnormalities, and serologic evidence of autoimmunity (anti-SSA or anti-SSB antibodies) [17]. Objective findings of ocular or oral dryness include tests such as the Schirmer test [17],[18]. Alternatively, ultrasonography demonstrating characteristic salivary gland abnormalities are also suggestive of SS [19].
While exocrine gland dysfunction leads to the typical presentation of oral and ocular dryness, SS has a wide spectrum of clinical presentations due to extra-glandular organ involvement [20]. Among the various extraglandular organ involvement, studies have shown that pulmonary findings are significant in many SS patients. An estimated 41–50% of patients with SS experience a cough with associated dyspnea [21]. Although imaging of such patients shows significant variability, cases of interstitial lung disease, pulmonary nodules, cystic changes, bronchial airway thickening, bronchiectasis, ground glass opacities, and consolidation have all been reported [21].
Sjögren’s syndrome was first considered after initial workup by pulmonology when labs revealed a positive RF and Sjögren SSA antibody (Table 1). Although SSA antibodies are not sufficient to make the diagnosis, SS was taken into consideration due to the patient’s respiratory symptoms mimicking that of pulmonary involvement in SS. Additionally, various features overlapped with regard to imaging. Such features include bronchiectasis, ground glass opacities, and consolidation (Figure 1 and Figure 2). Nevertheless, these pulmonary findings are nonspecific, and SS should not be diagnosed solely based on positive antibodies. With the patient lacking supportive features of ocular or oral dryness, SS is unlikely. If these characteristic symptoms develop, additional workup with Schirmer test or ultrasound may be indicated for further analysis.
Eosinophilic granulomatosis with polyangiitis
The American College of Rheumatology has defined six criteria to make the diagnosis of EGPA. These criteria include a history of asthma, CBC with a differential of >10% eosinophilia, mono or polyneuropathy, non-fixed pulmonary infiltrates, paranasal sinus abnormality, and blood vessel biopsy demonstrating extravascular eosinophils. The presence of four or more of these criteria corresponds to a specificity of 99.7% and sensitivity of 85% [22].
When considering the natural course of the presented patient’s disease, it characteristically matches that of EGPA. In fact, the patient presented with an initial prodromal phase characterized by asthma, followed by peripheral eosinophilia (Table 2), and finally a vasculitic phase with positive ANCA (Table 1). Furthermore, the patient met the following four diagnostic criteria: history of asthma, CBC with >10% eosinophils, non-fixed pulmonary infiltrates, and biopsy demonstrating extravascular eosinophils. With late signs of vasculitis and ANCA positivity, the patient did not initially meet the diagnostic criteria. However, as the disease characteristically progressed, the diagnosis of EGPA was finally made.
Management
One possible treatment for EGPA includes initiating high dose corticosteroids and tapering to a lower dose as the patient clinically improves. Additionally, the combination of azathioprine and daily steroids can help to maintain remission [23]. Low dose steroid use can also help manage the patient’s asthma symptoms that accompany EGPA [24].
In this case, the patient’s continued progression through the three phases of EGPA forced the physicians to adapt their treatment plan as the diagnosis developed. The asthma stage persisted for around 20 years and the accompanying symptoms were treated as needed with rescue and long-term inhalers. Throughout the 20 years, periodic lung infiltrates also occurred, and antibiotics and steroids were given to fight a presumed bacterial pneumonia. During these treatments, the steroids more likely had a larger effect than the antibiotics as the pulmonary infiltrates may have been caused by the persistent peripheral blood eosinophilia that can occur during the asthmatic phase of EGPA [25]. It was not until two days before the presentation of this case when the extremely elevated blood eosinophil count (81%) and pulmonary infiltrates turned the pulmonologist’s and eventually the hospitalist’s attention to treating eosinophilic pneumonitis. At this point, 40 mg methylprednisolone sodium intravenous twice daily was administered in the hospital, and the patient’s eosinophilia returned to normal limits (0%). The patient was discharged and told to continue 40 mg of prednisone daily. Follow-up appointments with the pulmonologist and a CT chest without contrast (Figure 3) two months after the presentation showed complete resolution of symptoms and of the previously identified areas of consolidation and ground-glass opacities throughout the lungs with no residual infiltrates or pleural effusions. Pulmonology started patient on a taper dose of 30 mg prednisone daily until the next visit, along with a referral to the rheumatologist for further investigation of the persistent positive ANA titer of 91 and positive SSA antibody with no peripheral signs of rheumatoid arthritis or Sjögren’s syndrome. The rheumatologist repeated an ANCA screen which came back with a new positive c-ANCA, the last diagnostic criteria needed to diagnosis EGPA. The rheumatologist recommended the patient continue 10 mg prednisone daily and start on 100 mg azathioprine tablet daily, which the patient tolerated with no medication side effects. Additionally, the patient had no significant ongoing wheezing or shortness of breath while using her rescue bronchodilator less than twice a week.
The patient continues to be followed closely by her primary care physician, rheumatologist, and pulmonologist to monitor additional possible outcomes of EGPA that can include repeat episodes of pulmonary infiltrates and nodules along with peripheral nerve damage, myalgias, hypertension, myocarditis, venous thromboembolism, lymphadenopathy, glomerulonephritis, and gastroenteritis [25].
Conclusion
Physicians should consider a wide range of conditions when evaluating pulmonary infiltrates. While EGPA is a very rare condition, it must remain in the physician’s differential due to its slowly progressive nature that can transition into a highly destructive disease without proper surveillance and treatment.
REFERENCES
1.
Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11. [CrossRef]
[Pubmed]

2.
Pagnoux C, Guillevin L. Churg-Strauss syndrome: Evidence for disease subtypes? Curr Opin Rheumatol 2010;22(1):21–8. [CrossRef]
[Pubmed]
3.
Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore) 1999;78(1):26–37. [CrossRef]
[Pubmed]
4.
Curtis C, Ogbogu PU. Evaluation and differential diagnosis of persistent marked eosinophilia. Immunol Allergy Clin North Am 2015;35(3):387–402. [CrossRef]
[Pubmed]
5.
Bjerrum OW, Pelliniemi TT, Wadenvik H. Care program for the diagnosis and treatment of eosinophilia. Nordic MPD Study Group. 2018. [Available at: http://nmpn.org/index.php/guidelines/18-care-program-for-the-diagnosis-and-treatment-of-eosinophilia-3rd-version-may-2018/file]
6.
Sharma P, Sharma S, Baltaro R, Hurley J. Systemic vasculitis. Am Fam Physician 2011;83(5):556–65.
[Pubmed]
7.
Agarwal R, Chakrabarti A, Shah A, et al. Allergic bronchopulmonary aspergillosis: Review of literature and proposal of new diagnostic and classification criteria. Clin Exp Allergy 2013;43(8):850–73. [CrossRef]
[Pubmed]
8.
Glancy JJ, Elder JL, McAleer R. Allergic bronchopulmonary fungal disease without clinical asthma. Thorax 1981;36(5):345–9 [CrossRef]
[Pubmed]
9.
Muthu V, Sehgal IS, Prasad KT, et al. Allergic bronchopulmonary aspergillosis (ABPA) sans asthma: A distinct subset of ABPA with a lesser risk of exacerbation. Med Mycol 2020;58(2):260–3. [CrossRef]
[Pubmed]
10.
Jederlinic PJ, Sicilian L, Gaensler EA. Chronic eosinophilic pneumonia. A report of 19 cases and a review of the literature. Medicine (Baltimore) 1988;67(3):154–62. [CrossRef]
[Pubmed]
11.
Marchand E, Reynaud-Gaubert M, Lauque D, Durieu J, Tonnel AB, Cordier JF. Idiopathic chronic eosinophilic pneumonia. A clinical and follow-up study of 62 cases. The Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM”O”P). Medicine (Baltimore) 1998;77(5):299–312. [CrossRef]
[Pubmed]
12.
Jeong YJ, Kim KI, Seo IJ, et al. Eosinophilic lung diseases: A clinical, radiologic, and pathologic overview. Radiographics. 2007;27(3):617–37. [CrossRef]
[Pubmed]
13.
Marchand E, Cordier JF. Idiopathic chronic eosinophilic pneumonia. Orphanet J Rare Dis 2006;1:11. [CrossRef]
[Pubmed]
14.
Danel C, Israel-Biet, D, Costabel U, et al. The clinical role of BAL in rare pulmonary diseases. Eur Respir Rev 1991;2:83.
15.
Matsuse H, Shimoda T, Fukushima C, et al. Diagnostic problems in chronic eosinophilic pneumonia. J Int Med Res 1997;25(4):196–201. [CrossRef]
[Pubmed]
16.
Churg A. Recent advances in the diagnosis of Churg-Strauss syndrome. Mod Pathol 2001;14(12):1284–93. [CrossRef]
[Pubmed]
17.
Reichlin M, Scofield RH. Ro (SS-A) antibodies. In: Shoenfeld Y, Gershwin ME, Meroni PL, editors. Textbook of Autoantibodies. 2ed. Amsterdam: Elsevier; 2006. p. 783–8.
18.
Vitali C, Moutsopoulos HM, Bombardieri S. The European Community Study Group on diagnostic criteria for Sjögren’s syndrome. Sensitivity and specificity of tests for ocular and oral involvement in Sjögren’s syndrome. Ann Rheum Dis 1994;53(10):637–47. [CrossRef]
[Pubmed]
19.
Luciano N, Baldini C, Tarantini G, et al. Ultrasonography of major salivary glands: A highly specific tool for distinguishing primary Sjögren’s syndrome from undifferentiated connective tissue diseases. Rheumatology (Oxford) 2015;54(12):2198– 204. [CrossRef]
[Pubmed]
20.
Asmussen K, Andersen V, Bendixen G, Schiødt M, Oxholm P. A new model for classification of disease manifestations in primary Sjögren’s syndrome: Evaluation in a retrospective long-term study. J Intern Med 1996;239(6):475–82. [CrossRef]
[Pubmed]
21.
Kreider M, Highland K. Pulmonary involvement in Sjögren syndrome. Semin Respir Crit Care Med 2014;35(2):255–64. [CrossRef]
[Pubmed]
22.
Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990;33(8):1094–100. [CrossRef]
[Pubmed]
23.
Bosch X, Guilabert A, Espinosa G, Mirapeix E. Treatment of antineutrophil cytoplasmic antibody associated vasculitis: A systematic review. JAMA 2007;298(6):655–69. [CrossRef]
[Pubmed]
24.
Sinico RA, Bottero P. Churg-Strauss angiitis. Best Pract Res Clin Rheumatol 2009;23(3):355–66. [CrossRef]
[Pubmed]
25.
Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: A clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore) 1984;63(2):65–81. [CrossRef]
[Pubmed]
SUPPORTING INFORMATION
Acknowledgments
Thank you to Dr. Sivakumar Nagaraju, MD for his dedication to and patience with his students.
Author ContributionsTyler Everett Wilson - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Sarven Tersakyan - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Guaranter of SubmissionThe corresponding author is the guarantor of submission.
Source of SupportNone
Consent StatementWritten informed consent was obtained from the patient for publication of this article.
Data AvailabilityAll relevant data are within the paper and its Supporting Information files.
Conflict of InterestAuthors declare no conflict of interest
Copyright© 2020 Tyler Everett Wilson et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}